This story originally appeared on the Broad Institute’s website.
Since the invention of the microscope, scientists have been painstakingly inspecting the neurons and other cell types that make up the nervous system to unravel their roles in health and disease. In recent years, single-cell methods have allowed researchers to identify new brain cell types, but they also wanted to know where those individual cells are in the brain, because their location can influence their function.
Now, two teams of scientists at the Broad Institute of MIT and Harvard have mapped the mouse nervous system with unprecedented depth and scale using recently developed technology called spatial transcriptomics, which can reveal not just the gene activity of individual cells, but also their locations within tissues and organs.
The two atlases, described in Nature, delineate hundreds to thousands of cell types across the mouse brain, revealing surprising cellular diversity in understudied brain regions and offering more precise and comprehensive looks at brain structures than were possible before.
One study was led by senior authors Evan Macosko, an institute member at the Broad and associate professor and attending psychiatrist at Massachusetts General Hospital, and Fei Chen, a core institute member at the Broad and an assistant professor in the Department of Stem Cell and Regenerative Biology at Harvard University. The other study was led by senior authors Xiao Wang, a Broad core institute member, a Merkin Institute Fellow, and an assistant professor of chemistry at MIT, and Jia Liu, an assistant professor of engineering at Harvard University.
The studies are part of a package of 10 papers in Nature that take distinct, yet complementary, approaches to mapping the mouse nervous system at the single-cell level. The studies are from groups at the Broad, Allen Institute for Brain Science, the Salk Institute for Biological Studies, and other institutions that are part of the National Institutes of Health’s Brain Research Through Advancing Innovative Neurotechnologies® Initiative, or The BRAIN Initiative — Cell Census Network (BICCN). Together the papers describe the first complete cell type atlas of a whole mammalian brain.
The team led by Macosko and Chen took an unbiased approach to measure activity of all genes in the genome of individual cells throughout the mouse brain, and assigned the cells’ locations within the tissue. Their analysis uncovered an estimated 90 percent of all cell populations in the mouse brain. The scientists found most cellular diversity within relatively understudied subcortical areas of the brain, especially the midbrain, pons, medulla, and the hypothalamus. “We suspected the most diversity would be found in these areas, so we prioritized them in our profiling,” said Macosko. “A lot of the real nuts and bolts stuff that a brain is doing is in these basic areas, which have received very little attention compared to the cortex. Our results underscore the need to study them more deeply.” The researchers also discovered clues about cellular function and the potential roles of brain structures in disease.
“Efforts like these generate crucial resources for the neuroscience community, because the brain is so enormously complicated,” said Chen.
Macosko added, “Our atlas represents the culmination of a decade of work at the Broad. Fei and I developed the technology in our labs and used it to process the largest single-cell and spatial dataset ever generated, leading to the first comprehensive atlas of cell types in a mammalian brain.”
The package also includes the spatial, single-cell atlas of the mouse brain and spinal cord that was led by Wang and Liu and was first published online in September in Nature. Independently of BICCN, the team used a spatial transcriptomics technology they developed to analyze the expression of more than 1,000 genes and detail the locations of individual cells with unparalleled sub-cellular resolution. They identified hundreds of cell types, generated highly precise, molecularly defined tissue maps of the mouse brain and spinal cord, predicted the spatial single-cell expression profiles of more than 10,000 additional genes, and demonstrated their method’s utility in revealing which cell types and brain regions are accessible with gene-delivery viruses.
“Our atlas represents the largest spatial single-cell dataset our lab has analyzed to date,” said Wang. “This resource provides a foundation for future work to further explore the roles of various genes, cells, and tissue regions in health and disease.”
An unbiased, comprehensive atlas
In the study from the Macosko and Chen labs, the scientists worked together to develop their spatial transcriptomic approach and apply it across the entire mouse brain. They also relied on the expertise of members of the Broad’s Genomics Platform and the team that supports Hail, an open-source tool for scalable genomic analysis.
The researchers identified several thousand unique cell populations, and mapped their locations in the whole tissue with near-cellular resolution. They estimated that their analysis captured about 90 percent of cell types in the mouse brain, including a large diversity in underexplored areas of the brain. The researchers created an online browser to house and share their datasets with the scientific community.
The team analyzed the full transcriptome of cells from nearly 100 regions across the mouse brain using high-throughput single-nucleus RNA sequencing, the preferred approach for efforts to create a human brain atlas. This resulted in more than four million profiles of gene activity, which they clustered into nearly 5,000 unique cell populations, most of which were neuronal cells.
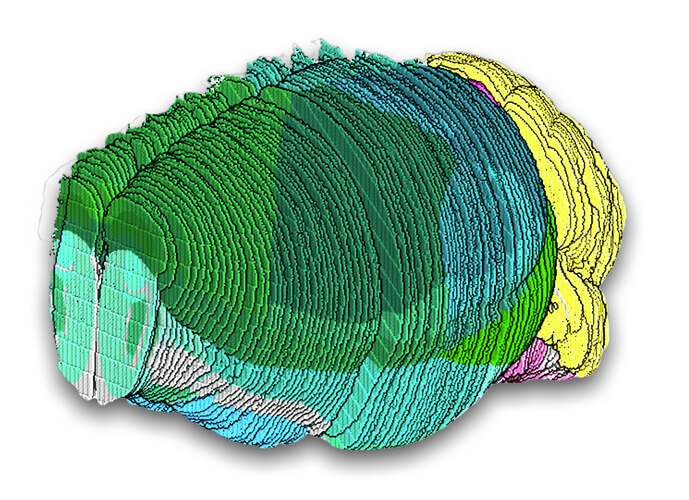
The team next applied an approach developed in the Chen and Macosko labs known as Slide-seq. They transferred 101 serial sections, spanning the volume of a single mouse brain, onto arrays of beads covered in unique DNA barcodes, which bound to the mRNA transcripts in the brain tissue. They then sequenced those transcripts and aligned that spatial data to an existing 3D reference atlas, enabling them to assign each transcript to a known brain structure, representing more than 1.7 million mapped cells. The researchers combined detailed and well-sampled cell type profiles from the single-nucleus sequencing dataset to locate each cell type in each slice to generate a detailed and thorough atlas of the entire mouse brain. They also analyzed the dataset to come up with two or three marker genes that could be used to uniquely identify almost all of the cell populations.
The authors said that other scientific groups can use the transcriptomic profiles, spatial localizations, and sets of marker genes they identified to study particular cells of interest. Groups may also use the data by integrating it with their own more detailed examination of a particular brain region. ”We hope that our atlas can both empower the community in their own work and allow them to further explore the cell types we identified,” said co-first author Jonah Langlieb, a computational biologist in the Macosko lab who led the work along with co-first author Nina Sachdev.
The team also revealed how signaling molecules known as neurotransmitters are used by different cell types in various brain regions. In addition, they demonstrated their atlas’s utility in revealing where disease-associated genes are active, for example, specific neuronal cells that are enriched for expression of genetic factors associated with schizophrenia.
(STAR)mapping a million cells
The team led by Wang and Liu used spatial transcriptomic tools they developed to characterize and map one million cells across the mouse brain and spinal cord. Through extensive molecular analysis and cellular annotation, the team produced maps pinpointing hundreds of different cell types. Clustering single cells based on gene expression patterns associated with various spatial niches in the brain allowed them to refine the boundaries of over a hundred tissue regions. The researchers also used their method to reveal which cell types and brain regions are accessible by harmless viruses called adeno-associated viruses (AAVs) that are engineered to deliver genes, demonstrating the value of this scalable approach for neuroscience research and the development of gene therapies. They shared their interactive atlas openly with the scientific community.
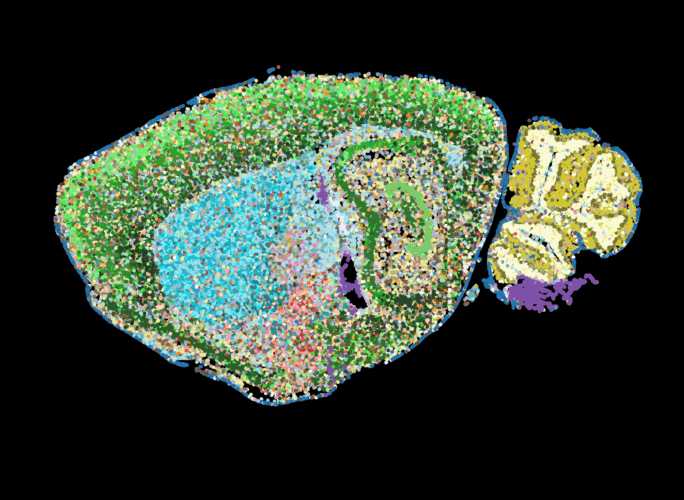
A series of detailed maps of the mouse nervous system feature colored dots indicating different cell types, which helped refine known tissue regions, shown here in a sample slice.
To generate the atlas, the research team employed a method, known as STARmap PLUS, which they previously developed to map gene expression of individual cells and their location in intact tissue samples.
The researchers measured the expression level of more than 1,000 brain-relevant genes in more than one million individual cells in the mouse brain and spinal cord to determine their identity. To do this, they treated tissue slices with molecular probes to detect and amplify specific mRNAs in circular structures that generate tight balls of DNA fragments from the bound mRNA, known as DNA nanoballs, and chemically treated the tissue to anchor the DNA in place. Using imaging-based in situ sequencing, the team was able to measure the spatial locations of these mRNAs at their native locations in the nervous system, with a subcellular resolution.
The researchers then plotted each cell’s type and location back onto a spatial map of each tissue slice. By comparing gene expression patterns between neighboring cells and examining other single-cell atlases, they were able to refine the anatomical boundaries of various tissue regions in the mouse nervous system. Their analysis identified 231 molecularly defined cell types and 106 tissue regions within the mouse brain and spinal cord.
“Through extensive annotation work, we analyzed the cells’ molecular features in the context of the previously established anatomical information about the brain,” said co-first author Hailing Shi, a postdoctoral research fellow in the Wang lab who led the work along with co-first authors Yichun He of Harvard University and Yiming Zhou, also a postdoctoral researcher in the Wang lab. “We wanted to bridge our molecular-rich data with the brain anatomical and tissue histological features that biologists are familiar with.”
The researchers also integrated existing single-cell datasets to impute, or predict, the expression of more than 10,000 additional genes in each cell at a lower cost than experimentally measuring all of those genes.
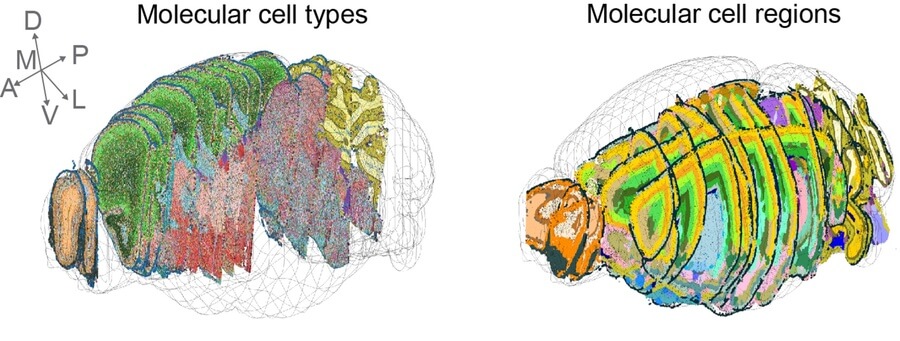
By registering images of brain tissue slices into a consensus coordinate framework, the researchers reconstructed cell and tissue composition in three dimensions.
In collaboration with Ben Deverman’s lab at the Broad, the team demonstrated how their method can potentially contribute to gene therapy development, by measuring how efficiently viral vectors deliver their gene cargo to specific cell types and tissue regions in the brain. In their study, the researchers focused on a variant of an engineered recombinant adeno-associated virus (rAAV) known as PHP.eB. The virus can cross the blood-brain barrier, but there was no systematic and quantitative knowledge about how well this vector delivers its cargo to particular cell types and brain subregions. After introducing barcode sequences into the PHP.eB viral genome, the researchers were able to use STARmap PLUS to measure the efficiency of the vector in reaching individual cells. Those data are also part of the team’s atlas.
As the scientific community generates and shares more single-cell datasets, the researchers plan to keep reevaluating their data and refining the maps in their atlas. They are also optimizing their experimental and computational pipelines to generate atlases of other mouse organs, in addition to those of non-human primates, and to continue applying their approach to study illnesses such as Alzheimer’s disease.
Together, these new atlases provide rich and complementary views of the remarkable complexity of the brain, and their datasets and methods set the stage for larger efforts to do the same for other mouse organs and for the human brain.
“By mapping the mouse brain, the primary mammalian system used in neuroscience, we’ve provided molecular, functional, and anatomical classifications that will be crucial to fully mapping the cells of the human brain, which is ultimately what we are most excited to do,” said Macosko.
FUNDING
The research from the Macosko and Chen labs was funded by the NIH BRAIN Initiative and by the Stanley Center for Psychiatric Research.
The Wang lab’s research was funded in part by the Searle Scholars Program, Thomas D. and Virginia W. Cabot Professorship, Edward Scolnick Professorship, Ono Pharma Breakthrough Science Initiative Award, and NIH DP2 New Innovator Award.
PAPERS CITED
Shi, H., He, Y., Zhou, Y. et al. Spatial atlas of the mouse central nervous system at molecular resolution. Nature 622, 552–561 (2023).
Langlieb, J., Sachdev, N., et al. The molecular cytoarchitecture of the adult mouse brain. Nature. December 13, 2023. DOI: 10.1038/s41586-023-06818-7.
