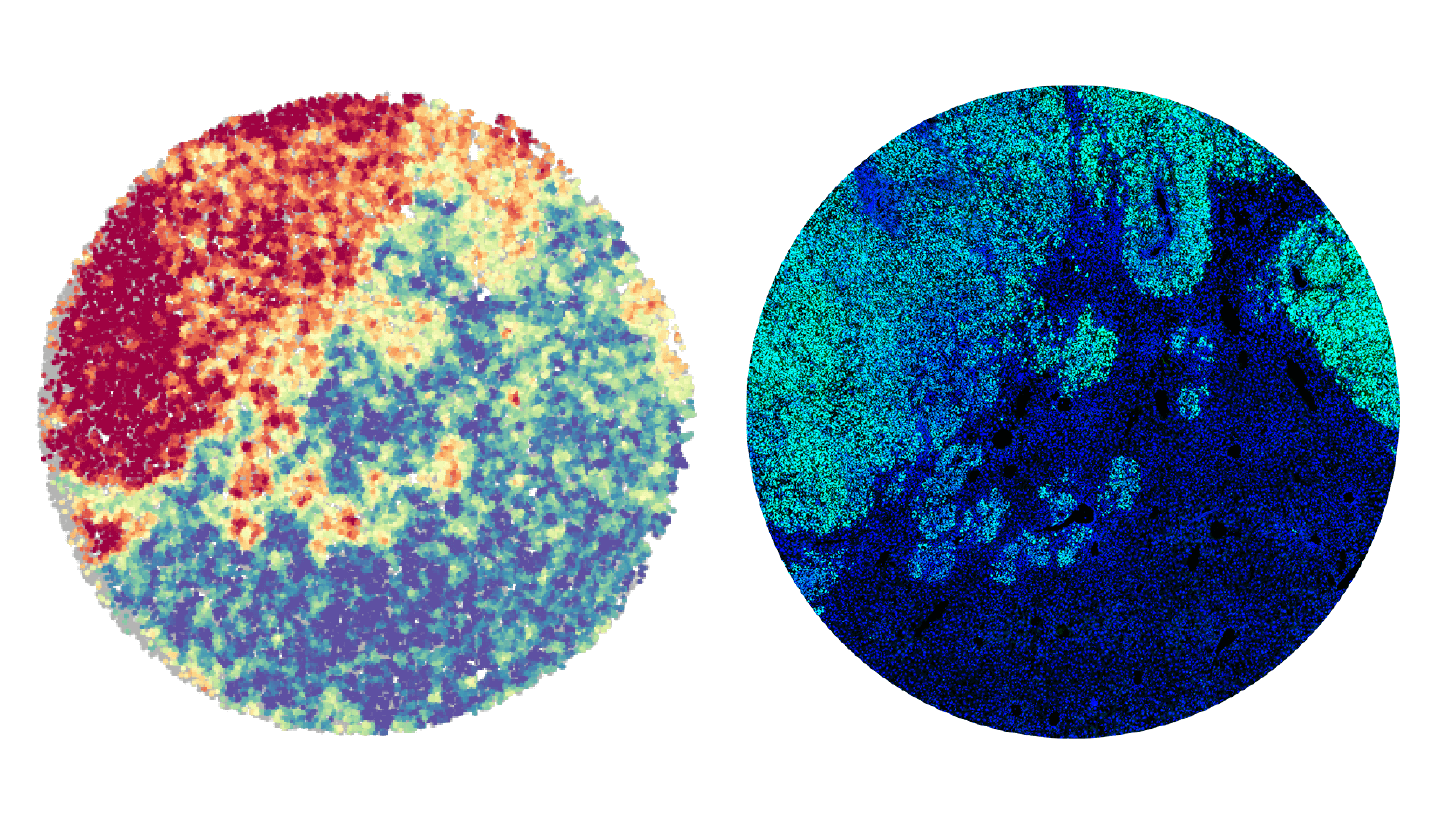
Within complex tissues such as cancer tumors, individual cells can vary widely from each other. Internally, cancer cells can develop unique DNA mutations and genomic changes, potentially leading to drug resistance, metastasis, or relapse. Externally, the cells’ specific locations within the tissue also matter, since the local structure of a tumor and its surrounding tissues can affect cell state and drug permeability.
To measure both genetic and local factors at the same time, researchers from Harvard University’s Department of Stem Cell and Regenerative Biology (HSCRB) and the Broad Institute of MIT and Harvard developed a new technique of spatially resolved DNA sequencing, called “slide-DNA-seq.” When further combined with spatially resolved gene expression analysis, the technology gives researchers a better understanding of cancer progression and potential treatment.
The researchers’ findings are published in the journal Nature.
The slide-DNA-seq approach involves analyzing slices of intact tissue, where each cell is kept in its original location — as opposed to conventional sequencing techniques, where cells are dissociated before DNA extraction. The researchers used microbeads, each tagged with a unique spatial barcode, to capture DNA from the tissue. “Each bead is about the size of a cell. Together, they are like individual pixels on a camera which takes snapshots of genomic alterations within each cell in the tissue,” said co-corresponding author Fei Chen, who is an assistant professor in HSCRB and core institute member at the Broad Institute.
After measuring genomic changes, the researchers applied a related RNA-based Slide-seq method that Chen and collaborators developed in 2019, which uses gene expression profiles to identify and map the locations of cell types and subtypes within a tissue slice. “We can use the same barcoded beads to capture the transcriptome from each cell, and combine these two measurements into a multi-modal image,” Chen said. In other words, the researchers could see changes in both the cells’ DNA and gene expression within the context of tissues.
The researchers tested the combined technique to a mouse model of metastatic cancer, as well as a primary tumor sample from a patient with colorectal cancer. In each tissue, the researchers were able to identify subpopulations of cancer cells in distinct regions, corresponding with unique genomic evolutionary paths and gene expression states.
“We applied this to DNA sequencing and cancer, which are hugely important areas of medical research,” said co-corresponding author Jason Buenrostro, who is a HSCRB assistant professor and associate member of the Broad Institute. “However, the technology also opens the door to more broadly measuring DNA molecules within tissues. As such, we imagine future approaches will use this work as a foundation to build tools that measure modifications to DNA as well, i.e., the epigenome.”
In this article


This study received funding from the Allen Institute Distinguished Investigator Award, the National Institutes of Health, a National Human Genome Research Institute training grant, the Burroughs Wellcome Fund and the Harvard Quantitative Biology Initiative.
Source article: Zhao, T., Chiang, Z. D., et al. (2021). Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature. DOI: 10.1038/s41586-021-04217-4